리뷰 날짜: 2024.07.18
January 2023
Zerun Lin, Le Ou-Yang
https://academic.oup.com/bib/article/24/1/bbac586/6965907
Preliminary
Siamese Neural Network

- 구조가 유사한 두 네트워크들로 구성
- weight을 공유함
- training 과정
- 두 개의 input에 대한 embedding을 구함
- 두 embedding 사이의 거리를 구함
- 두 입력이 같은 클래스에 속하면 거리를 가깝게, 다른 클래스에 속하면 거리를 멀게 학습시킴
- 장점
- 소량의 데이터만으로 학습 가능 (one-shot learning을 위해 개발되었으므로)
- embedding으로 변환한 후에 별도의 classifier를 붙여 feature extractor로 사용 가능
Contrastive Learning (Contrastive Loss)

- self-supervised learning
- 유사한 이미지가 임베딩 공간에서 서로 가깝게, 다른 이미지는 서로 멀리 있도록 저차원 공간에서 이미지를 인코딩하는 방법을 모델이 학습하는 것
- contrastive loss: 벡터 사이의 유사성을 정량화하는 방법으로, 모델이 올바른 임베딩을 학습하도록 유도함

-
- Y: 두 데이터의 label (유사하면 0, 유사하지 않으면 1)
- D: 데이터 간의 거리 (주로 L2거리 사용)
- m: 임계값 (유사하지 않은 데이터 간의 최소 거리)
- positive pair: 유사한 데이터, Y=0일 때 D를 최소화하도록 학습
- negative pair: 유사하지 않은 데이터, Y=1일 때, D가 m 이상이 되도록 학습
Attention Mechanism

- 딥러닝 모델에서 중요한 정보를 더 집중적으로 학습하도록 하는 매커니즘
- 입력 문장의 모든 단어를 동일한 가중치로 취급하지 않고, 출력 문장에서 특정 위치에 대응하는 입력 단어들에 더 많은 가중치를 부여
- 입출력의 길이가 달라도, 모델이 더 정확하고 유연하게 학습할 수 있음
- attention function을 사용해 가중치를 계산함
VGG (Visual Geometry Group) network
- 신경망의 깊이가 딥러닝의 정확도에 큰 영향을 미친다는 것을 보여준 모델
- 이미지 분류 작업에서 높은 성능을 보임
- 단순한 3x3 convolution 필터로 깊은 네트워크를 구성
- 깊은 구조로 인해 많은 메모리, 계산 자원이 필요하고 훈련 시간이 길다는 단점이 있음
Backbone
- 신경망의 핵심 구조
- CNN에서는 이미지의 특징을 추출하기 위해 사용되는 기본 네트워크를 의미
Sigmoid function

- 결과값으로 항상 0과 1의 사이의 값을 반환
- 이상치가 들어와도 0이나 1에 수렴하므로 이상치 문제를 해결하며 연속된 값을 전달함
- 이 때문에 활성화 함수로 sigmoid를 사용하면 이진 분류를 할 수 있음
Introduction
GRNs (Gene Regulatory Networks)
- TFs와 target genes 사이의 causal regulatory relationship으로 구성되어, 전사 (transcription) 과정과 세포가 어떻게 행동할지를 결정함
- *** TF (Transcription Factor): transcription을 촉진/억제해서 gene expression을 조절하는 단백질
- 대량 유전자 발현 데이터로부터 GRNs를 재구성하기 위해 많은 계산법이 제안되었음
- unsupervised learning 기반 inference: 단순히 gene expression data를 사용해 유전자 간의 regulatory relationship을 추론함
- E.g., correlation-based (pearson/spearman’s correlation 사용해서 유전자 간의 co-expression levels를 캡처함), Gaussian graphical-model-based (유전자 간의 직접적인 상호작용을 추론할 수 있음)
- scRNA-seq 기술이 발전함에 따라 single cell resolution의 gene expression data가 축적됨
- single cell 수준에서 GRNs를 추론할 수 있게 되었음
- 그러나 scRNA-seq 데이터에는 높은 noise rate과 dropout events라는 문제가 있음
- *** dropout: scRNA-seq에서의 dropout은 유전자가 발현되었지만, 검출되지 않는 현상을 의미 (= technical false zero)
-
최근에는 TF-gene interaction data가 축적되어 GRN 추론을 위해 supervised learning 기반 network 추론 방법이 제안되었음
-
supervised learning은 알려진 유전자 regulatory interaction을 활용해서 gene expression level과 gene regulatory interaction 간의 correlation을 학습할 수 있음 -> unsupervised learning보다 우수한 성능
-
E.g., DeepSEM (GRN과 representations of scRNA-seq data를 함께 추론하는 deep generative model), GRGNN (로 유전자 발현 데이터로부터 GRNs를 재구성한 graph neural network model), CNNC (histogram image를 사용해 유전자 쌍의 co-expression 관계를 나타내는 CNN 기반 모델)
-
-
supervised learning 기반의 방법들은 single data source로부터 GRN을 추론하도록 설계되어 있음
-
최근에 scRNA-seq 기술이 발전하며 동일한 cell type의 gene expression data를 다른 플랫폼이나 시점에서 수집할 수 있음
-
이들을 통합하면 GRN을 더 정확하게 추론할 수 있지만 서로 다른 소스에서 수집하는 것에 다음과 같은 문제가 존재함
-
중복이 발생할 수 있음
-
gene regulatory interaction의 예측에 기여하는 정도가 다를 수 있어서, 동등하게 처리하면 잘못된 예측을 하게 됨
-
-> 여러 개의 single data source를 효과적으로 통합할 수 있는 새로운 GRN inference 방법이 필요함
DeepMCL (Deep Multi-View Contrastive Learning)
- 데이터 속성에 대한 가정을 하지 않는 일반화된 supervised learning 기반 network inference model
- supervised learning은 알려진 유전자 regulatory interaction을 활용해서 gene expression level과 gene regulatory interaction 간의 correlation을 학습할 수 있음 -> unsupervised learning보다 우수한 성능
- E.g., DeepSEM (GRN과 representations of scRNA-seq data를 함께 추론하는 deep generative model), GRGNN (로 유전자 발현 데이터로부터 GRNs를 재구성한 graph neural network model), CNNC (histogram image를 사용해 유전자 쌍의 co-expression 관계를 나타내는 CNN 기반 모델)
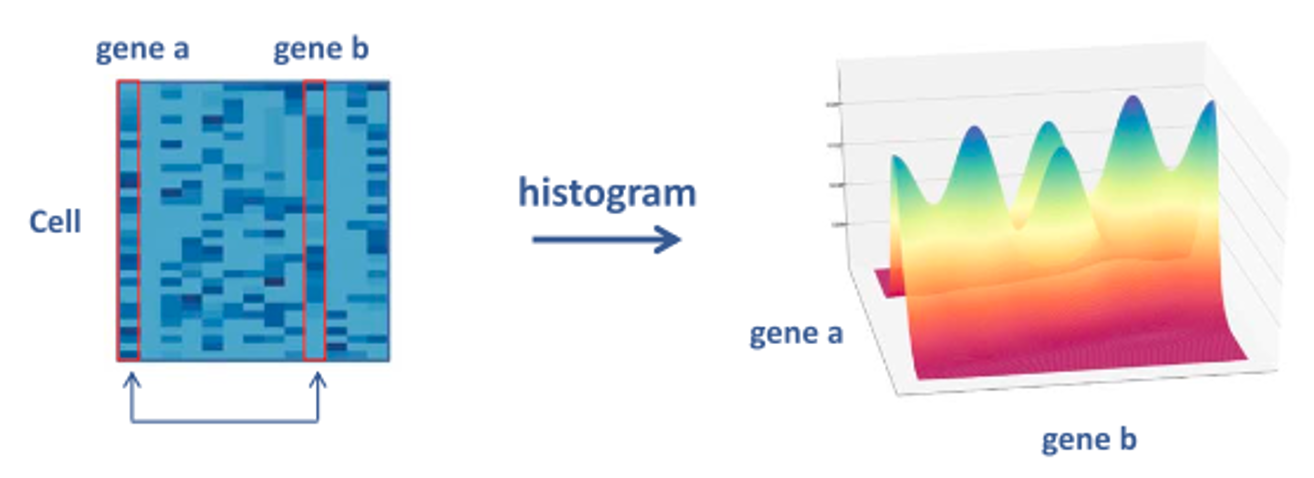
- 각 유전자 쌍을 histogram images로 나타냄 -> gene expression levels의 분포를 시각적으로 표현
- 여러 single data source에서 수집한 scRNA-seq data를 통합하기 위해 multi-view learning framework를 도입
- Siamese CNN과 contrastive loss를 도입해서 각 image의 embedding을 추출 -> positive (regulatory interaction이 있는) gene pairs와 negative (regulatory interaction이 없는) gene pairs 구분 가능
- attention module을 도입해서 다른 data sources와 neighbor gene pairs이 제공하는 정보를 효과적으로 통합
Methods
1. Representation of gene pairs
- 각 유전자 쌍을 histogram image set으로 변환해서 gene expression level의 distribution을 표현
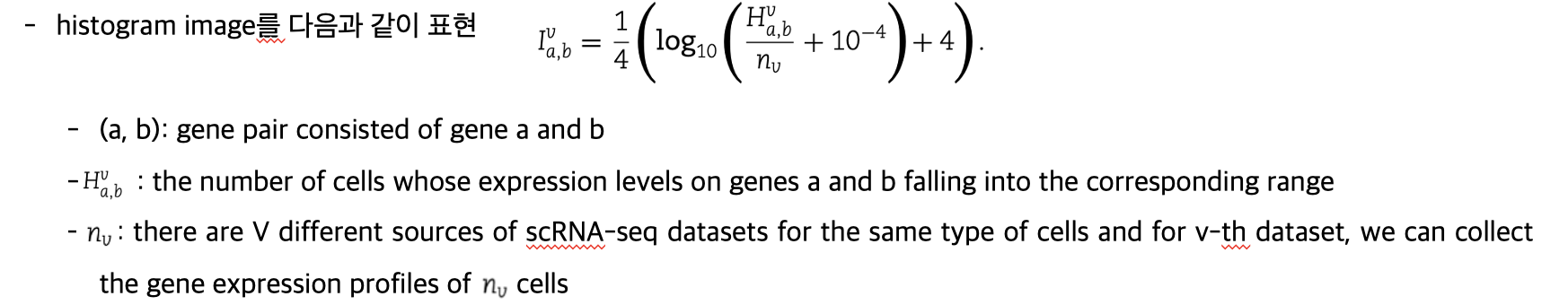
2. Generation of primary and neighbor images

3. Model Formulation
- 각 이미지에 대한 embedding을 개별적으로 학습하지 않고, contrastive learning을 사용
- positive gene pairs와 negative gene pairs를 구분하기 위해
- two-stage learning
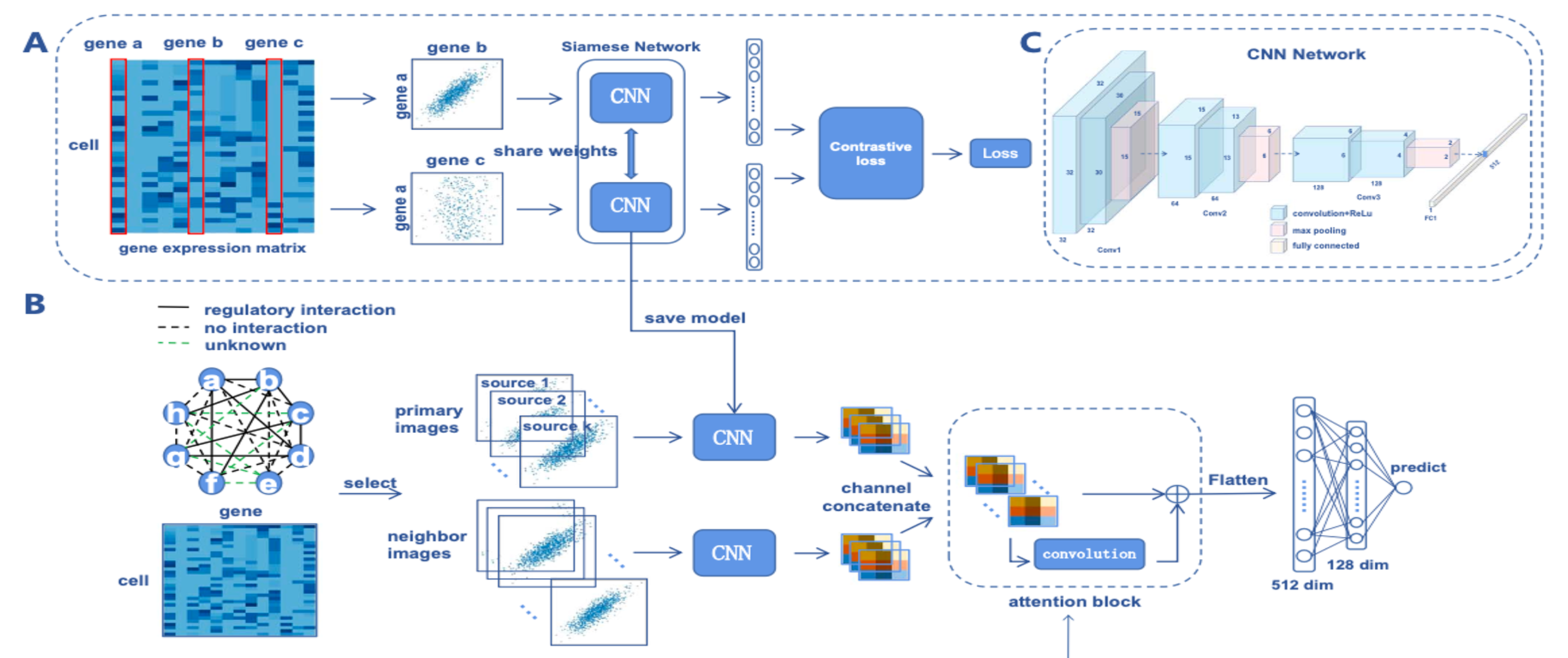
- 1st stage: 2개의 동일한 CNN 모델이 있는 Siamese Convolutional Neural Network와 contrastive loss를 사용해서 positive/negative gene pairs의 embedding 추출
- VGG network와 같은 classic CNN architecture를 backbone model로 사용해서 gene interaction과 관련된 패턴 학습
-
2nd stage: 학습된 CNN network를 사용해 primary와 neighbor image embedding을 추출하고, 다음 예측을 수행
-
non-local block attention module을 사용해 모든 image embedding을 통합 (예측에 대한 image contribution이 다르므로)
-
attention의 output은 flatten되어 2개의 fully connected layers에 연결되고, binary classification을 위해 sigmoid 함수 사용
-
Simulation Studies
Experiment results on synthetic data
- DeepMCL과 DeepDRIM, 각 데이터셋을 개별적으로 분석하는 축소된 버전의 DeepMCL(DeepMCL-) 및 DeepDRIM의 두 가지 변형(attention modules을 포함한 DeepDRIM+A와 대비 학습 모듈을 포함한 DeepDRIM+C)과 비교하기 위한 ablation studies를 실시
***ablation studies: machine learning system의 building blocks을 제거해서 전체 성능에 미치는 효과에 대한 insight를 얻기 위한 과학적 실험
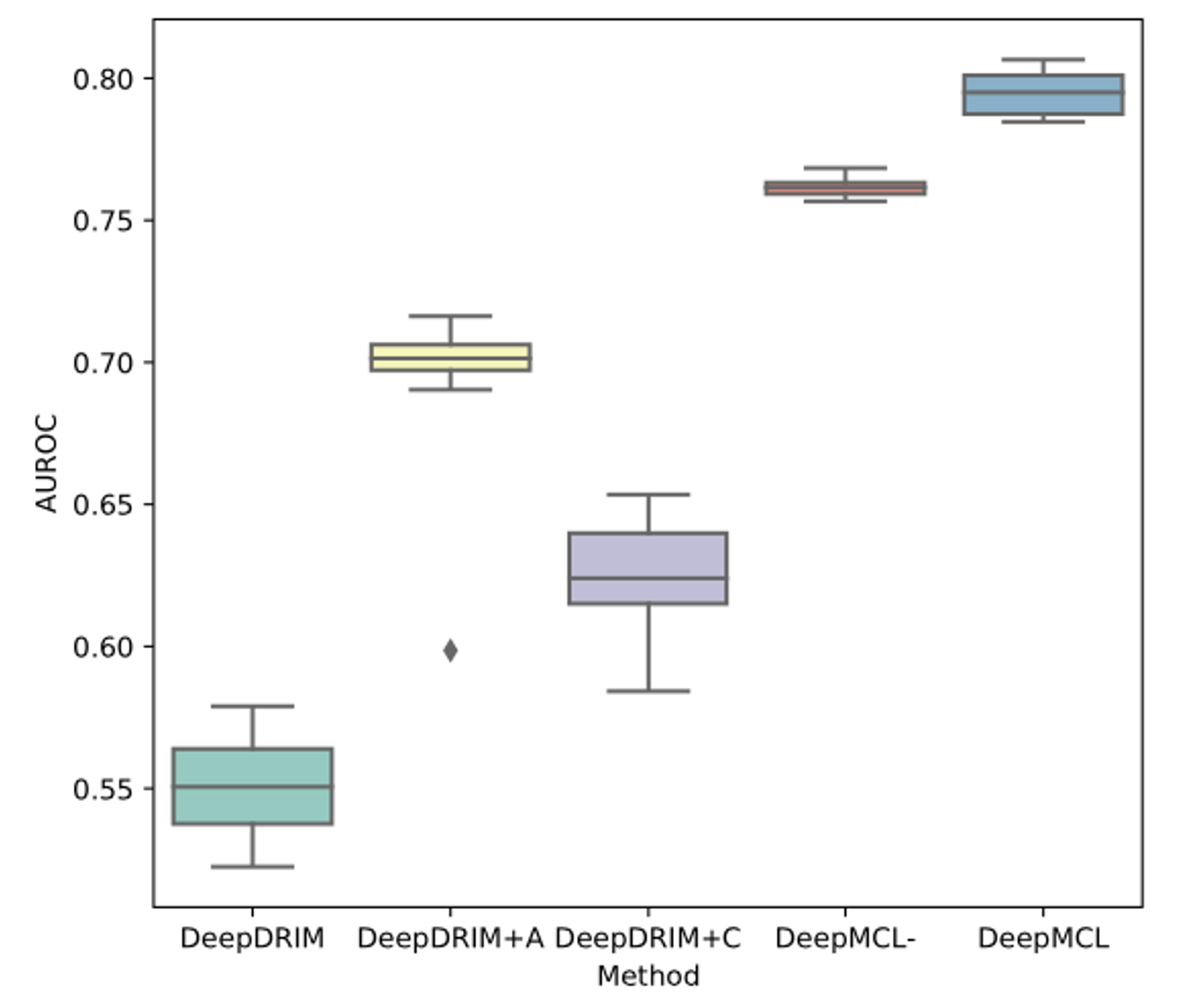
- DeepDRIM과 나머지 버전의 성능 비교
- DeepDRIM+A의 성능 향상: attention module의 효과를 입증, 모든 이웃 이미지가 예측에 기여하지 않는다는 것을 의미
- DeepDRIM+C의 성능 향상: 대비 학습 모듈의 효과를 입증, positive gene pairs의 embedding을 negative gene pairs의 embedding과 구별할 필요가 있음을 의미
- DeepMCL-의 성능이 DeepDRIM+A와 DeepDRIM+C보다 우수함, contrastive learning과 attention module을 통합한 효과
-> DeepMCL은 DeepMCL-보다 우수하고, 여러 데이터 소스를 통합하는 이점이 있음
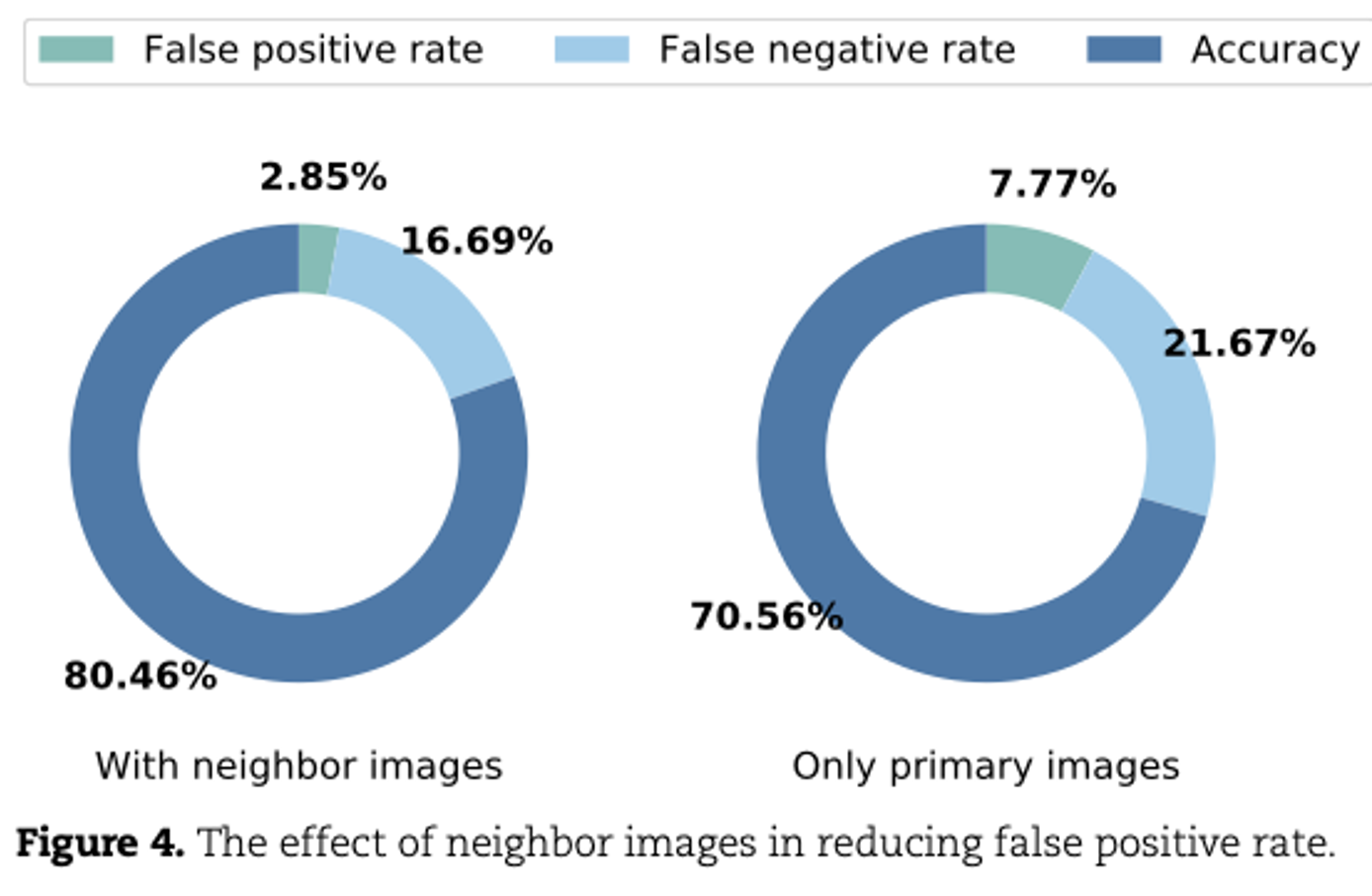
Effect of neighbor images in reducing false positive
- 유전자 상호작용을 추론할 때 neighbor images를 고려하는 이점을 확인하기 위해, 두 가지 다른 합성 데이터 세트로 DeepMCL을 훈련시킴
- 첫 번째 세트는 primary images만 포함하고, 두 번째 세트는 neighbor images도 포함하고, 같은 validation set로 훈련시킴
- neighbor genes의 영향을 고려하면 FP를 크게 줄일 수 있고, FN은 감소하고, 정확도는 10% 증가함
Effect of Image Size
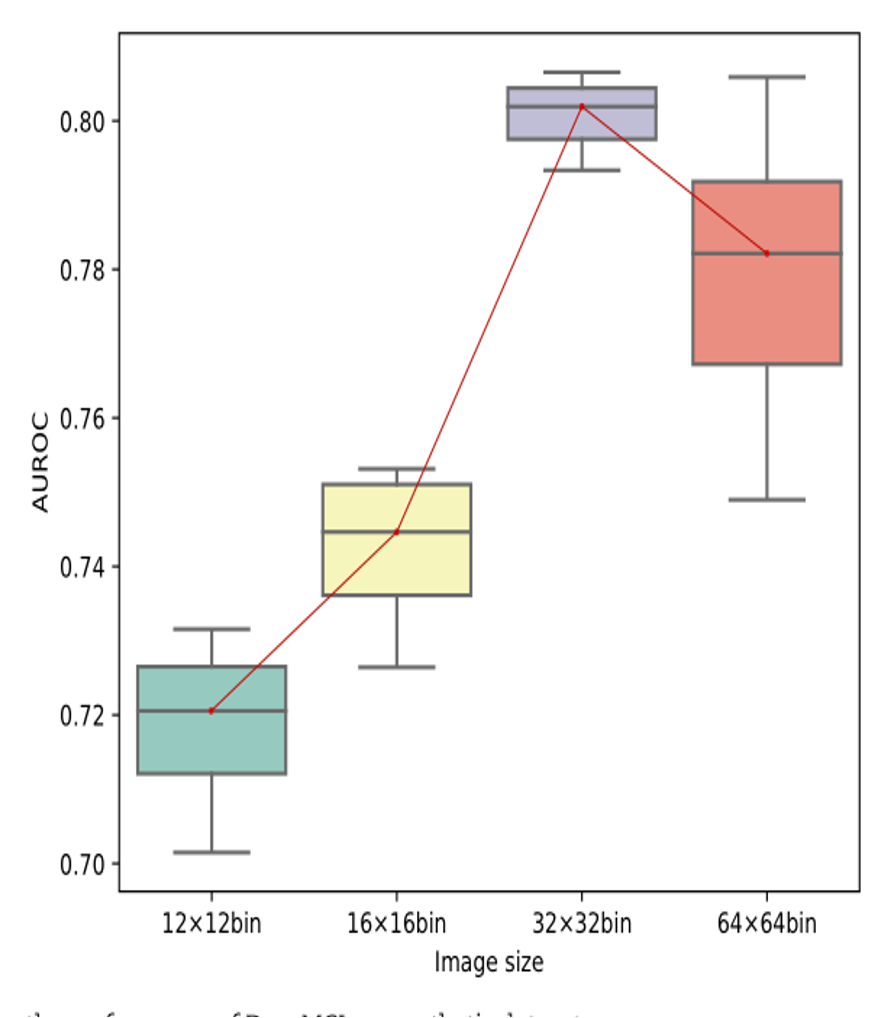
- 히스토그램의 작은 이미지 크기는 정보 손실을 초래할 수 있는 반면, 큰 이미지 크기는 과도한 노이즈를 포함할 수 있음 -> 이미지 크기가 DeepMCL의 성능에 미치는 영향을 평가하는 실험 수행
- 위의 합성 데이터를 기반으로 히스토그램 이미지를 생성할 때 네 가지 다른 이미지 크기(12 × 12, 16 × 16, 32 × 32, 64 × 64)를 고려
- DeepMCL의 성능은 이미지 크기가 증가함에 따라 증가함
- 32 × 32에서 최상의 성능에 도달한 후 감소
- 작은 이미지 크기는 유용한 정보를 잃을 수 있고, 큰 이미지 크기는 일부 노이즈 정보를 도입할 수 있기 때문으로 추측
- 이후의 실험에서는 이미지 크기를 32 × 32로 고정함
Real Data Analysis
Application to mHSC scRNA-seq datasets
- 3종류의 mHSC (쥐의 조혈모세포) lines의 scRNA-seq 데이터 사용
- 적혈구 계통(mHSC(E)), 과립구-대식세포 계통(mHSC(GM)) 및 림프구 계통(mHSC(L))
- 각 데이터에 대해 random한 개수의 TF-gene interaction pairs를 positive sample로 random하게 선택하고, 같은 개수의 non-interacting TF-gene pairs를 negative sample로 선택함 (실험에서는 18개를 선택함)
- 세 데이터셋에 다양한 모델을 적용하여 TF-유전자 상호작용 예측에서의 성능을 평가
- DeepMCL을 최신의 다섯 가지 유전자 네트워크 추론 모델(PIDC, SINCERITIES, SCODE, CNNC, DeepDRIM)과 비교
- PIDC, SINCERITIES, SCODE는 unsupervised learning model이고, CNNC와 DeepDRIM은 supervised learning model
- 3-fold cross-validation AUC score를 사용하여 모델의 성능을 평가

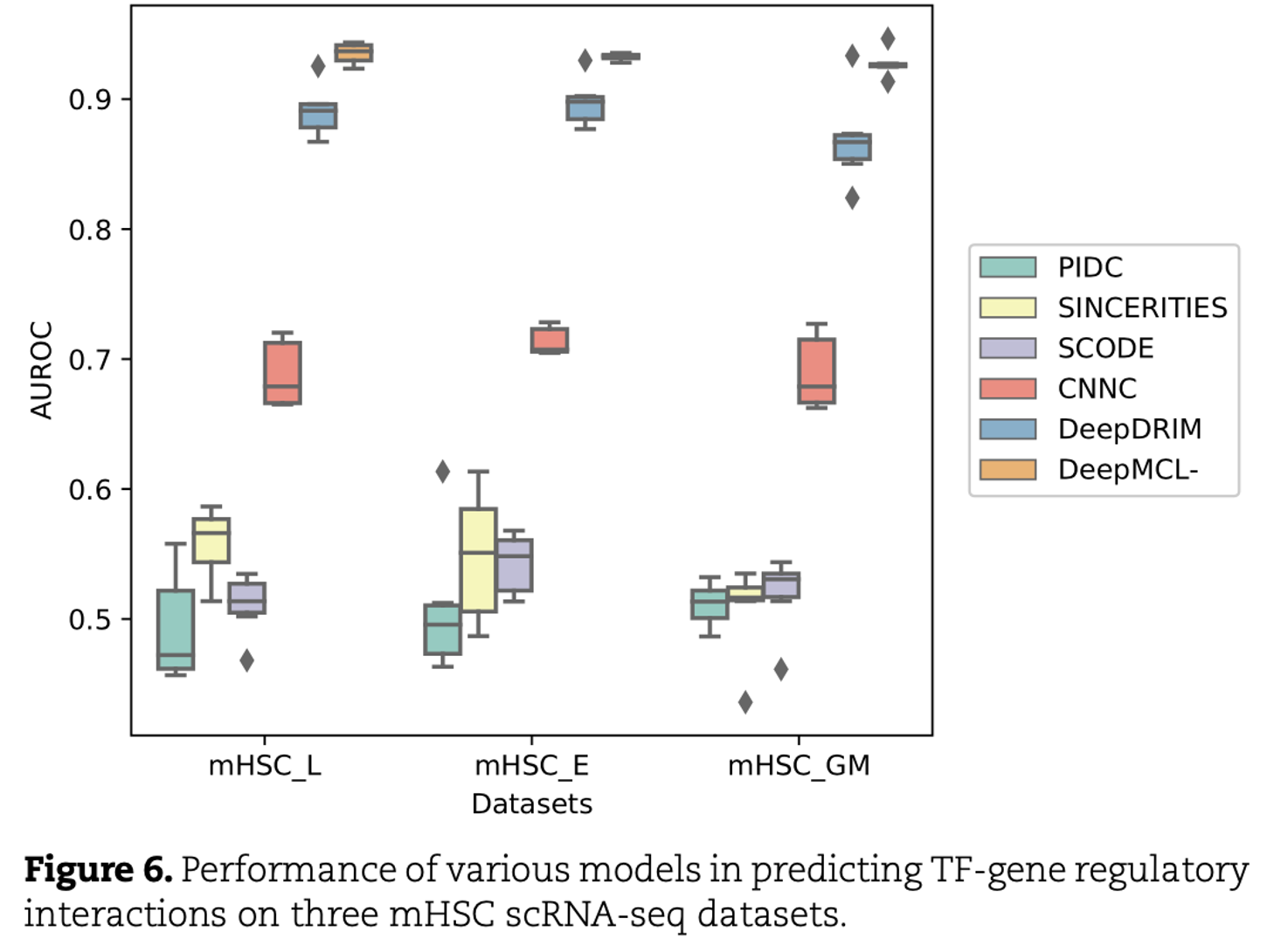
- supervised learning model이 모든 데이터셋에서 unsupervised learning model보다 우수함
- DeepMCL-은 모든 데이터셋에서 DeepDRIM과 CNNC보다 우수한 성능을 보이고, attention module과 contrastive learning module을 도입한 이점을 입증
- DeepMCL은 단일 데이터셋 분석에서 가장 우수한 성능을 보이고, TF-gene interactions 예측에서 모델의 효과를 보임
Effect of data scale
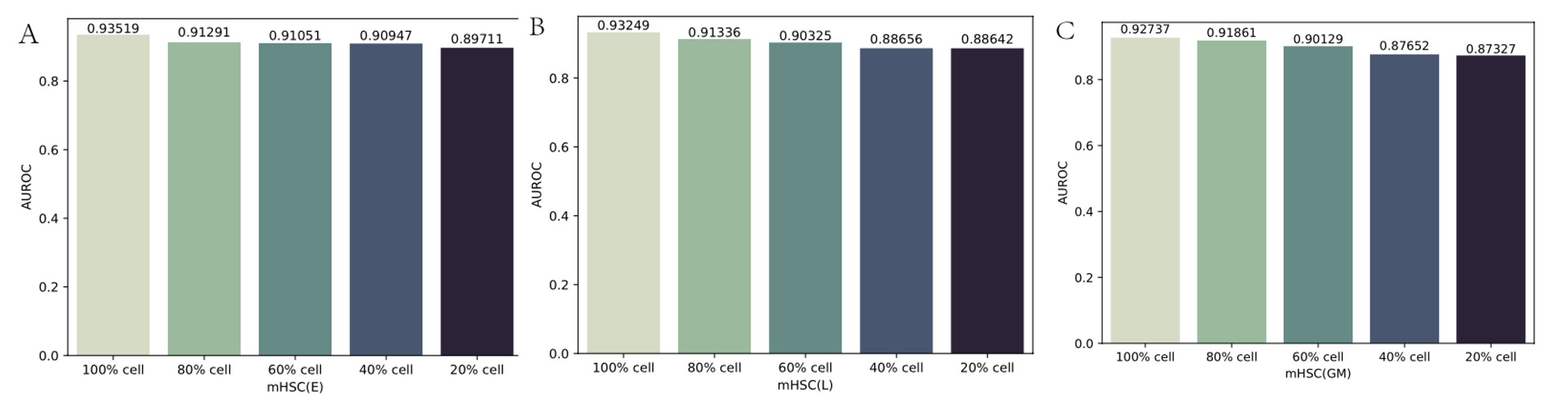
- 세포 수가 감소할 때 DeepMCL의 성능 변화를 평가하기 위해 downsampling 실험을 수행
- 원래 데이터셋의 세포 비율을 random sampling하여 세포의 하위 집합을 생성하고 이 하위 집합을 사용하여 각 gene pair의 primary와 neighbor image를 계산했고, 원래 데이터셋의 80%, 60%, 40%, 20%의 비율을 고려하여 다른 세포 하위 집합을 생성함
- 하위 집합 크기가 감소함에 따라 DeepMCL의 성능이 약간 감소하지만, 원래 데이터셋의 20%의 세포만 사용하여 TF-gene interaction을 예측하더라도 DeepMCL은 여전히 좋은 성능을 발휘함
- 유전자 조절 상호작용을 추론하는 데 있어 DeepMCL의 효과와 견고성을 입증
Application to time-course scRNA-seq data
- static data보다 time-series data가 gene regulatory interactions를 추론하기에 적합
- mESC (쥐의 배아줄기세포)와 hESC (인간 배아줄기세포)의 2개의 time-series scRNA-seq 데이터셋을 사용
- DeepMCL과 3개의 gene network inference model TDL-3D CNN, TDL-LSTM, dynGENIE3을 비교
- TDL-3D CNN, TDL-LSTM -> supervised learning model
- dynGENIE3 -> unsupervised learning model
- 3-fold cross validation으로 모델 성능을 평가
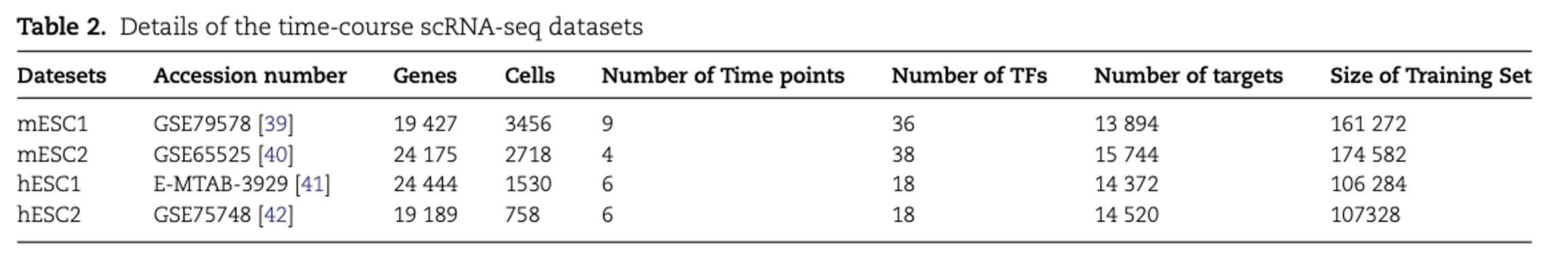
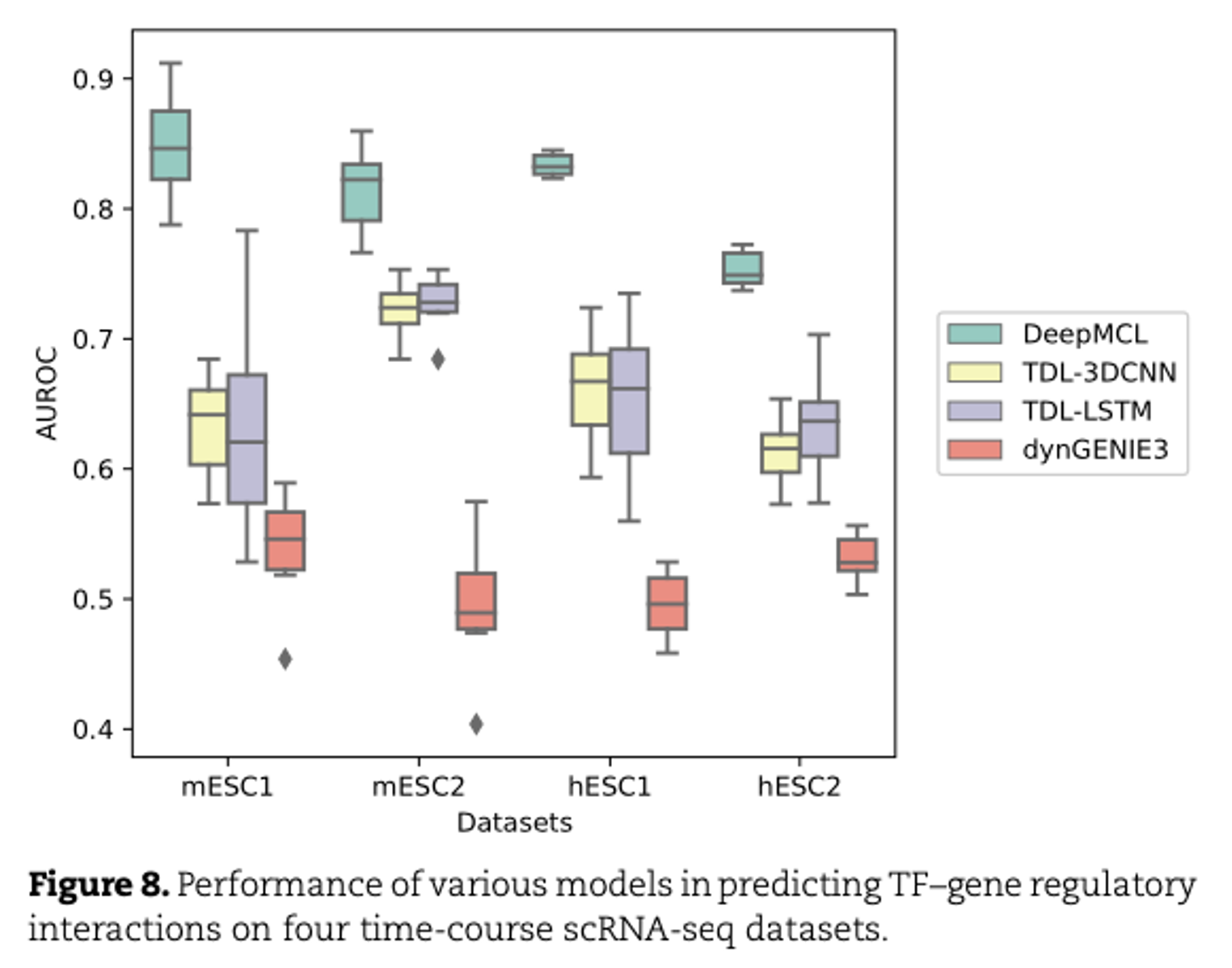
- dynGENIE3이 가장 낮은 성능을 보임 (unsupervised learning model이므로)
- DeepMCL은 모든 데이터셋에서 가장 뛰어난 성능을 보이고, time-series dataset에서 gene network를 추론하는 데 있어 DeepMCL이 효과가 있음을 의미
Discussion
- 논문에서는 여러 플랫폼이나 시점에서 수집된 gene expression data를 통합해서 single GRN을 추론하는 데 초점을 둠
- 이미 알려진 gene regulatory interaction에 의존하는 supervised learning 모델임
- 실제로는 network를 알 수 없을 때도 있고, GRN의 구조도 세포의 상태에 따라 변경될 수 있음
- 세포 상태 별 network를 추론하고 훈련 세트에 대한 dependency를 줄여야 함